Factors affecting drug absorption span physicochemical properties of the molecule, pharmaceutical/formulation variables, and physiological/pathological factors at the site of administration, and together they determine the rate and extent of absorption (bioavailability) that drive onset and intensity of effect. Mastery of these determinants explains variability between patients, fed–fasted states, dosage forms, and routes, enabling rational selection of route and formulation for predictable therapeutic exposure.
Key definitions
Absorption is the movement of an unmetabolized drug from the site of administration into the systemic circulation, occurring by passive diffusion, carrier-mediated transport (active or facilitated), endocytosis, or via paracellular aqueous pores depending on the barrier and the drug’s properties. Bioavailability is the rate and extent to which the active drug reaches systemic circulation, and it is directly shaped by absorption processes and presystemic metabolism in the gut wall and liver.
Mechanistic backbone
For most small molecules, passive transcellular diffusion dominates and follows Fick’s principle where flux is proportional to permeability, surface area, and the concentration gradient across the membrane. Carrier-mediated uptake and efflux (e.g., OCT1 for metformin and P-glycoprotein in intestinal epithelium) introduce saturability, competition, and directionality that can enhance or restrict absorption depending on transporter expression and substrate affinity.
Ionization and pH
Weak electrolytes exist in ionized and unionized forms, and the unionized fraction typically permeates lipid membranes more readily; this fraction depends on the Henderson–Hasselbalch relationship and local pH. For weak acids, \( \text{fraction unionized} = \frac{1}{1 + 10^{pH - pKa}} \), and for weak bases, \( \text{fraction unionized} = \frac{1}{1 + 10^{pKa - pH}} \), which explains why acidic drugs may absorb more in acidic media and basic drugs in more alkaline segments, all else being equal.
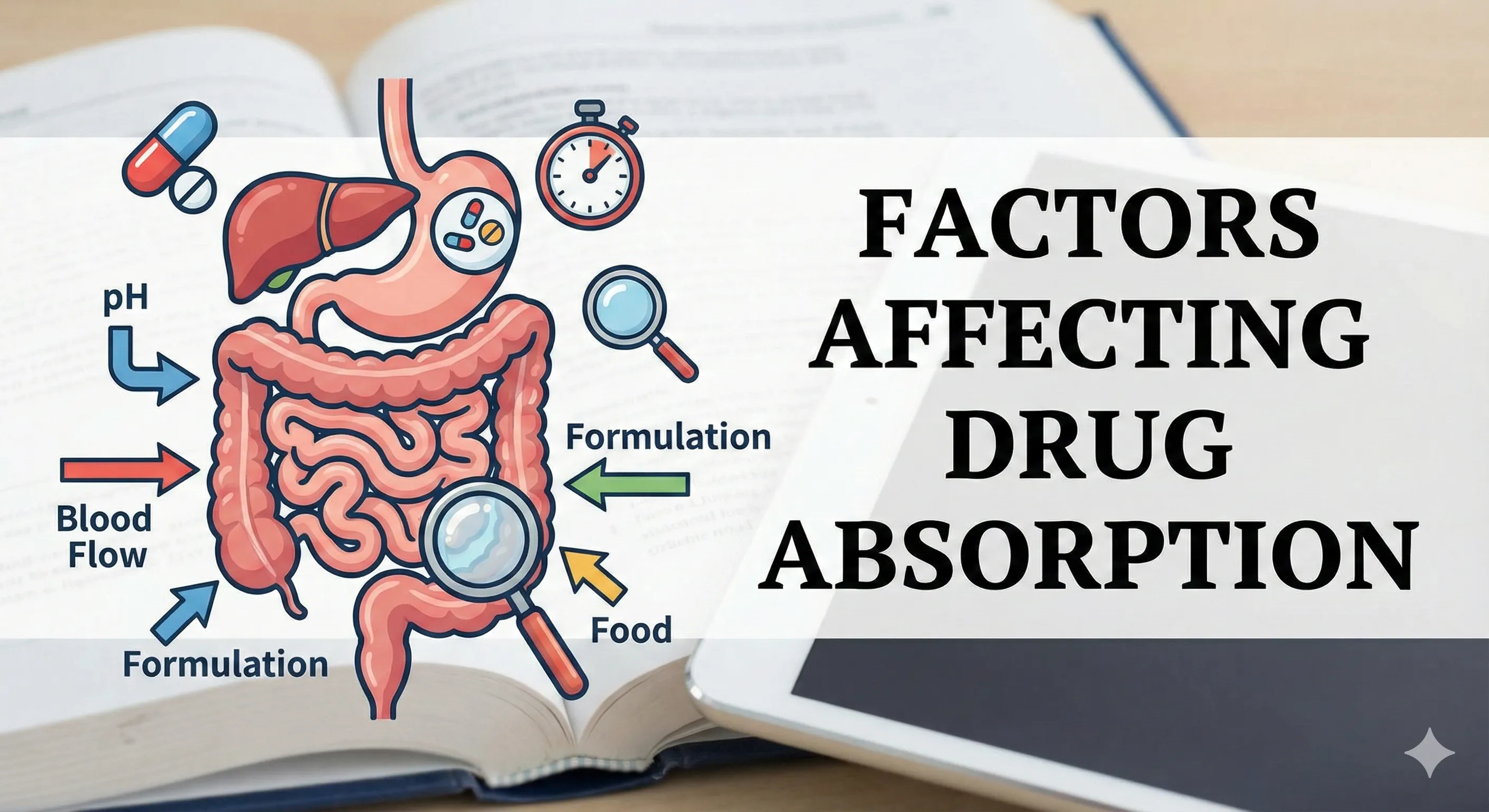
Physicochemical factors
- Lipid solubility (logP/logD) increases membrane permeability up to a point, whereas extreme lipophilicity can slow dissolution and limit effective concentration at the absorption surface.
- Molecular size inversely correlates with passive permeability; smaller molecules diffuse more readily across lipid bilayers than larger, polar structures.
- Ionization (pH–pKa) governs the unionized fraction and thus passive diffusion, with local pH shifts altering absorption along the GI tract or across other epithelia.
- Aqueous solubility and dissolution rate are rate-limiting for solid dosage forms; micronization increases surface area and dissolution, often improving absorption for poorly soluble drugs.
- Solid-state form (polymorph vs amorphous) and salt selection can markedly change apparent solubility/dissolution and bioavailability.
Pharmaceutical/formulation factors
- Dosage form hierarchy for oral absorption generally favors solutions over suspensions over capsules over tablets, with enteric coating delaying release and modified-release systems flattening and extending input.
- Excipients (e.g., surfactants, disintegrants, complexing agents) can increase wetting and dissolution or conversely bind the drug and reduce free concentration.
- Controlled-release, enteric coatings, and multiparticulate systems alter dissolution site and rate, shaping Tmax and Cmax without changing intrinsic permeability.
Gastrointestinal physiology
- Surface area is maximal in the small intestine due to villi and microvilli, making it the dominant site of absorption for most orally administered drugs despite pH-based predictions favoring the stomach for weak acids.
- Gastric emptying rate is a major determinant of oral absorption rate because it controls delivery to the high-area small intestine; delayed emptying slows, and accelerated emptying hastens absorption.
- Intestinal motility influences contact time; hypomotility can promote complete dissolution/absorption for some formulations, whereas hypermotility can reduce uptake.
Splanchnic perfusion and gradients
- Blood flow at the absorption site maintains the concentration gradient by carrying absorbed drug away, so reduced perfusion (critical illness, shock) slows absorption kinetics.
- Exercise or warm conditions may increase local perfusion and slightly enhance absorption for some routes and sites.
Transporters and gut-wall metabolism
- Apical efflux transporters like P-glycoprotein can pump drug back into the lumen, lowering net absorption and interacting with inhibitors/inducers.
- Enterocyte enzymes (e.g., CYP3A, esterases, UGTs) contribute to presystemic loss (first-pass in the gut wall) before portal delivery to the liver, reducing bioavailability.
Food effects and luminal composition
- Food can delay gastric emptying and change luminal pH and bile secretion, variably decreasing Cmax for some drugs but enhancing absorption of lipophilic compounds that benefit from bile-mediated solubilization.
- A mechanistic PK model for fenofibrate demonstrated increased absorption in the fed state (especially high-fat meals), illustrating bile and GI handling effects on a lipophilic prodrug.
- Luminal complexation (e.g., tetracyclines with calcium in dairy) forms insoluble chelates that reduce effective absorption despite adequate permeability.
Pathophysiology and interindividual factors
- Ageing can reduce gastric acid secretion, slow gastric emptying, and alter intestinal perfusion and transport capacities, leading to changes in absorption rate and extent.
- GI pathology that reduces surface area (e.g., villous atrophy, surgical short gut) or increases inflammation/mucus can decrease uptake regardless of formulation.
- Critical illness redistributes blood flow away from the splanchnic circulation and impairs motility, generally reducing and destabilizing oral absorption.
Route-specific absorption considerations
- Pulmonary delivery leverages large alveolar surface area and thin barriers for rapid uptake; deposition depends on particle size, flow dynamics, and device technique.
- Transdermal systems must overcome the stratum corneum barrier; permeation improves with appropriate physicochemical profiles and can be enhanced with microneedles or iontophoresis.
- Nasal mucosa supports fast systemic absorption for suitable molecules but is limited by dose volume, mucociliary clearance, and local enzymatic activity.
Clinical implications
- When high first-pass metabolism limits oral bioavailability, selecting routes that bypass first-pass or using prodrugs and enabling formulations can restore systemic exposure.
- Understanding ionization and pH allows prediction of “ion trapping” across pH gradients (e.g., more basic compartments retaining weak acids), which can influence local and systemic distribution after absorption.
Decision framework for optimization
- Match drug properties (solubility, permeability, pKa) to formulation design (particle size, salt form, release profile) and dosing conditions (fed/fasted, timing) to control input rate and extent.
- Anticipate transporter/enzyme effects and food interactions, and modify co-medications or administration instructions to avoid avoidable absorption loss.
Core equations and conceptual anchors
- Passive diffusion aligns with Fick’s concept: absorption rate rises with permeability, area, and luminal concentration, falling with thicker or more resistant barriers.
- Henderson–Hasselbalch links fraction unionized to pH and pKa: for weak acids \( \log\frac{[A^-]}{[HA]} = pH - pKa \); for weak bases \( \log\frac{[B]}{[BH^+]} = pKa - pH \), guiding predictions of segmental absorption.
Table 1. Physicochemical factors affecting absorption
| Factor | Typical effect | Practical notes |
|---|---|---|
| Lipophilicity (logP/logD) | Increases transcellular diffusion until dissolution limits dominate | Balance solubility and permeability to avoid dissolution-limited uptake |
| Molecular size | Smaller molecules permeate faster | Large polar molecules may need carriers or non-oral routes |
| Ionization (pH–pKa) | Unionized fraction permeates membranes better | Segmental pH determines fraction unionized in GI tract |
| Solubility/dissolution | Faster dissolution increases effective luminal concentration | Micronization and salts improve dissolution-limited absorption |
| Solid state (polymorph/amorphous) | Alters solubility and dissolution profile | Select higher-solubility forms to increase bioavailability |
| Complexation/chelation | Reduces free drug for absorption | Avoid co-administration with chelating foods/ions |
Table 2. Pharmaceutical factors (oral)
| Variable | Effect on absorption | Examples/notes |
|---|---|---|
| Dosage form | Solutions > suspensions > capsules > tablets for onset | Enteric/modified release shift site/rate of input |
| Excipients | Surfactants/disintegrants increase dissolution; binders may reduce it | Formulation-specific instruction may be required |
| Particle size | Smaller size increases surface area and dissolution | Micronized digoxin shows high bioavailability |
| Coatings | Enteric protects from acid, delays to intestine | Useful for acid-labile or irritant drugs |
Table 3. GI physiological determinants
| Determinant | Typical influence | Clinical remark |
|---|---|---|
| Surface area (intestine) | Major driver of extent of absorption | Villous atrophy/short gut reduce exposure |
| Gastric emptying | Major driver of rate (Tmax) | Fatty meals delay, prokinetics accelerate |
| Motility/transit | Alters contact time and dissolution | Diarrhea reduces, ileus may increase absorption |
| Perfusion | Maintains gradient; low flow slows uptake | Shock reduces and destabilizes absorption |
| pH and bile | Modulate ionization and solubilization | Bile aids lipophilic drug solubilization |
| Transporters/enzymes | Efflux/metabolism lower net absorption | P-gp/CYP3A in enterocytes reduce bioavailability |
Table 4. Food effects and interactions
| Situation | Mechanism | Consequence |
|---|---|---|
| High-fat meal with lipophilic drug | Bile-mediated solubilization and altered emptying | Higher absorption/Cmax/AUC for some drugs |
| Dairy with tetracyclines | Calcium chelation reduces free drug | Lower absorption and efficacy |
| Altered gastric pH | Ionization/dissolution changes | Variable, drug-specific fed–fasted differences |
Route-specific absorption contexts
- Lungs: Efficient uptake with appropriate aerodynamic diameter; deposition heterogeneity and technique are key to delivered dose and systemic absorption where intended.
- Skin: Stratum corneum is rate-limiting; chemical enhancers, microneedles, or energy-based methods increase flux for suitable molecules without first-pass loss.
Putting it into practice
- If dissolution limits absorption, reduce particle size, choose a more soluble salt or polymorph, or use enabling formulations (e.g., surfactant systems).
- If first-pass and efflux limit exposure, consider sublingual, transdermal, nasal, or parenteral routes, or use transporter/enzyme inhibitors where clinically appropriate and safe.
High-yield takeaways
- Absorption is a joint function of permeability, dissolution, surface area, perfusion, transporters, and presystemic metabolism; modifying any one of these can change bioavailability and onset.
- Fed–fasted instructions, spacing from chelating foods, and formulation selection are practical levers to stabilize exposure in daily prescribing.
References
- Alagga AA, Pellegrini M, Gupta V. Drug Absorption. In: StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
- Ritter JM, Flower R, Henderson G, Loke YK, MacEwan D, Robinson E, Fullerton J. Rang & Dale’s Pharmacology. 10th ed. London: Elsevier; 2024. Chapter 9: Absorption and distribution of drugs.
- Factors which influence gastrointestinal drug absorption. Deranged Physiology; 2023.
- Martinez MN, Amidon GL. A mechanistic approach to understanding the factors affecting drug absorption: a review of fundamentals. J Clin Pharmacol. 2002;42(6):620–43.
- Fen Y, Choi YH, Kim M-G, et al. A mechanism-based pharmacokinetic model of fenofibrate for explaining increased drug absorption after food consumption. BMC Pharmacol Toxicol. 2018;19(1):7.
- Patton JS, Byron PR. Pulmonary drug delivery. Part I: physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol. 2003;56(6):588–99.
- Vora LK, Donnelly RF, Larrañeta E, et al. Strategies for improving transdermal administration: new approaches to controlled drug release. Pharmaceutics. 2023;15(4):1183.
- Rowland M, Tozer TN. The ABCD of clinical pharmacokinetics. Br J Clin Pharmacol. 2013;76(1):17–19.